Discovering versatile biocatalysis that can be used to accelerate the synthesis of drugs and agrochemicals.
Based in the Frick Chemistry Laboratory at Princeton University, the Hyster lab integrates the fields of organic synthesis, organometallic chemistry, chemical biology, and protein engineering to develop new biocatalytic reactions that solve longstanding selectivity challenges and expand how enzymes can be used to achieve sustainable chemical synthesis.
Recent publications.
-

Engineering a photoenzyme to use red light
Publication Abstract
Photoenzymatic reactions involving flavin-dependent “ene”-reductases (EREDs) rely on protein-templated charge transfer (CT) complexes between the cofactor and substrate for radical initiation. These complexes typically absorb in the blue region of the electromagnetic spectrum. Here, we engineered an ERED to form CT complexes that absorb red light. Mechanistic studies indicate that red-light activity is due to the growth of a red-absorbing shoulder off the previously identified cyan absorption feature. Molecular dynamics simulations, docking, and excited-state calculations suggest that the cyan feature involves a π→π∗ transition on flavin, whereas the red-light absorption is a π→π∗ transition between flavin and the substrate. Differences in the electronic transition are due to changes in the substrate-binding conformation and allosteric tuning of the electronic structure of the cofactor-substrate complex. Microenvironment tuning of the CT complex for red-light activity is observed with other engineered photoenzymatic reactions, highlighting this effect’s generality.
-
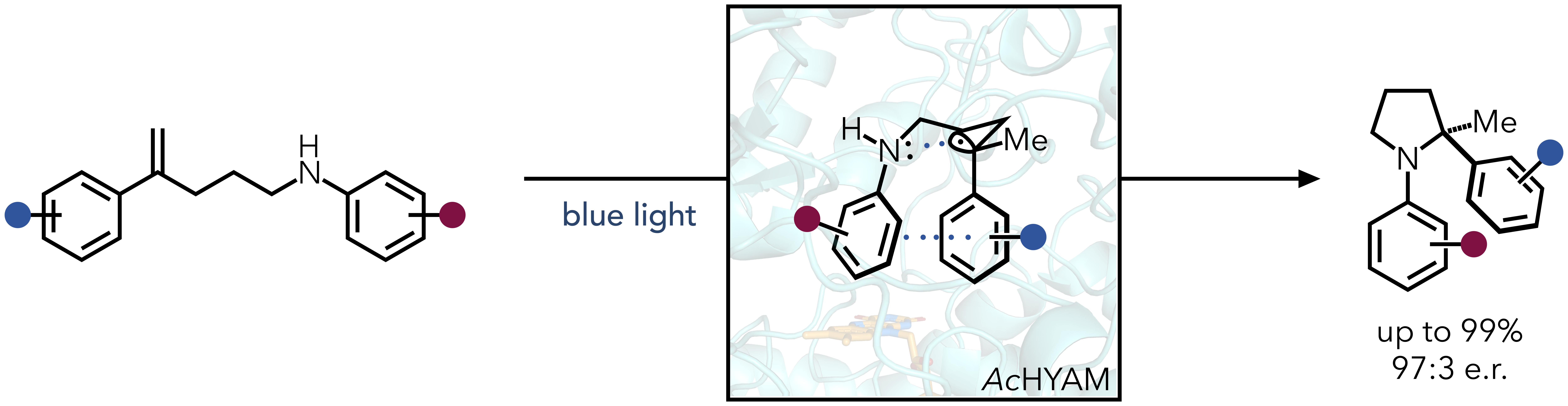
Emergence of a distinct mechanism of C–N bond formation in photoenzymes
Publication Abstract
C–N bond formation is integral to modern chemical synthesis due to the ubiquity of nitrogen heterocycles in small-molecule pharmaceuticals and agrochemicals. Alkene hydroamination with unactivated alkenes is an atom economical strategy for constructing these bonds. However, these reactions are challenging to render asymmetric when preparing fully substituted carbon stereocenters. Here, we report a photoenzymatic alkene hydroamination to prepare 2,2-disubstituted pyrrolidines by a Baeyer-Villiger Monooxygenase. Five rounds of protein engineering afforded a mutant, providing excellent product yield and stereoselectivity. Unlike related photochemical hydroaminations, which rely on the oxidation of the amine or alkene for C–N bond formation, this work exploits a through-space interaction of a reductively generated benzylic radical and the nitrogen lone pair. This antibonding interaction lowers the oxidation potential of the radical, enabling electron transfer to the flavin cofactor. Experiments indicate that the enzyme microenvironment is essential in enabling a novel C–N bond formation mechanism with no parallel in small molecule catalysis. Molecular dynamics simulations were performed to investigate the substrate in the enzyme active site which further support this hypothesis. This work is a rare example of an emerging mechanism in non-natural biocatalysis, where an enzyme has access to a mechanism that its individual components do not. Our study showcases the potential of enhancing emergent mechanisms using protein engineering to provide unique mechanistic solutions to unanswered challenges in chemical synthesis.
-
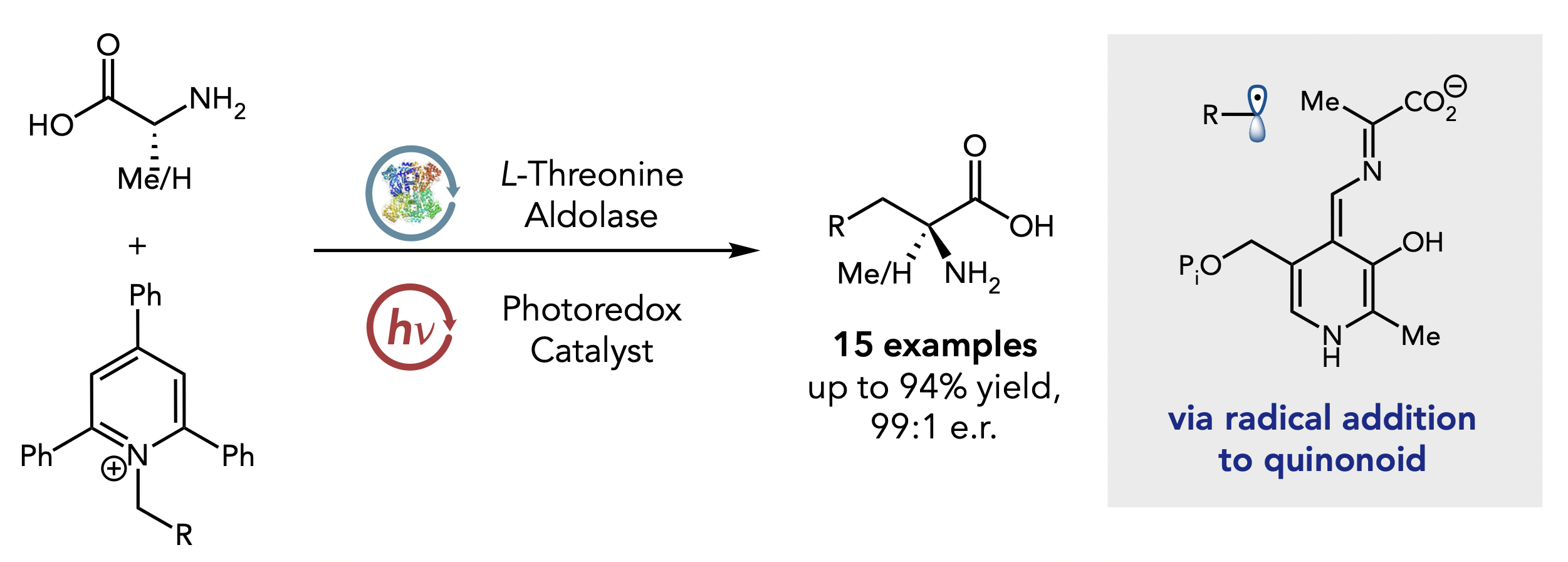
Synergistic Photoenzymatic Catalysis Enables Synthesis of a-Tertiary Amino Acids Using Threonine Aldolases
J. Am. Chem. Soc. 2024, 146, 20, 13754–13759.
Publication Abstract
a-Tertiary amino acids are essential components of drugs and agrochemicals, yet traditional syntheses are step-intensive and provide access to a limited range of structures with varying levels of enantioselectivity. Here, we report the α-alkylation of unprotected alanine and glycine by pyridinium salts using pyridoxal (PLP)-dependent threonine aldolases with a Rose Bengal photoredox catalyst. The strategy efficiently prepares various a-tertiary amino acids in a single chemical step as a single enantiomer. UV-vis spectroscopy studies reveal a ternary interaction between the pyridinium salt, protein, and photocatalyst, which we hypothesize is responsible for localizing radical formation to the protein active site. This method highlights the opportunity for combining photoredox catalysts with enzymes to reveal new catalytic functions for known enzymes.

Our team.
The Hyster Lab is led by Principal Investigator, Todd Hyster. Our research team comprises a group of exceptional post-doctoral associates and fellows, graduate students, and undergraduates.